





























Next-generation sequencing (NGS) has transformed genomics research by enabling simultaneous analysis of hundreds or thousands of samples in a single run. This is made possible with indexed adapters containing short nucleotide sequences or "barcodes" that label individual libraries. However, an often-overlooked challenge in multiplexed sequencing experiments is index hopping: the misassignment of reads to the wrong sample due to unintentional transfer of index sequences between libraries. Although the proportion of affected reads is small, ranging from 0.1% to 2% on some Illumina platforms, the impact can be profound in sensitive applications.
Understanding Index Hopping
Index hopping occurs when an index sequence from one library becomes erroneously associated with a different library fragment. On Illumina platforms that utilize patterned flow cells and exclusion amplification (ExAmp) chemistry, such as the HiSeq® 4000, NovaSeq® 6000, and NextSeq® 2000, this issue is especially pronounced. In ExAmp®, DNA fragments and amplification primers coexist in solution rather than being surface bound, increasing the likelihood that free-floating adapters can anneal to and amplify unintended fragments. This leads to the propagation of reads tagged with incorrect index combinations, which in turn results in misassigned sequences that can pass standard quality filters.
Residual adapter contamination during library preparation is a significant contributor to this issue. Incomplete removal of indexing primers or ligated adapters can create a pool of free-floating barcodes that readily participate in cross-sample annealing during cluster generation. High cluster density, short insert sizes, and AT-rich sequences can further exacerbate the likelihood of hopping by promoting strand displacement or inefficient adapter ligation.
Prevalence of Index Hopping on Illumina platforms
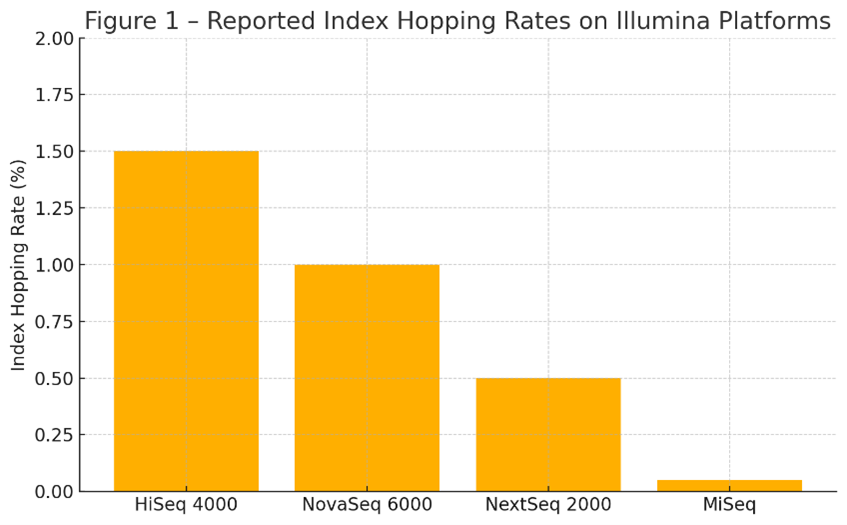
The rate of index hopping varies by instrument and library quality. Patterned flow cell instruments using ExAmp® chemistry, including the HiSeq® 4000 and NovaSeq® 6000, routinely exhibit index hopping rates between 0.1% and 2%. The NextSeq® 2000, although improved, can still demonstrate rates approaching 0.5% under certain conditions. In contrast, the MiSeq® platform, which uses bridge amplification and unpatterned flow cells, typically shows hopping rates below 0.05% due to its more confined clustering environment.
Figure 1 – Reported Index Hopping Rates on Illumina Platforms
This figure highlights the importance of indexing strategy selection, especially for high-throughput workflows. A 1% misassignment rate in a 1-billion-read NovaSeq® run equates to 10 million reads placed in the wrong sample, a critical concern for any study relying on low-allele-frequency detection or absolute quantification.
Consequences of Index Hopping
The implications of index hopping are especially severe in precision applications. In oncology-focused panels, index hopping can generate false-positive low-frequency variants by transferring reads with genuine mutations into unrelated samples. Similarly, in microbiome and metagenomic studies, index hopping introduces non-native taxa into the read pool, skewing diversity and abundance analyses.
Single-cell and spatial transcriptomic assays are also highly sensitive to index misassignment. A single hopped read bearing the wrong barcode can alter cluster definitions, increase apparent doublet rates, and reduce interpretability. Moreover, in clinical sequencing environments governed by CAP or CLIA standards, sample cross-talk must be kept to less than 0.2%.
Unique Dual Indexing: A robust solution to Index Hopping
Unique dual indexing (UDI) is the most effective strategy for mitigating index hopping in Illumina workflows. In a UDI setup, each library is tagged with a unique combination of i7 and i5 index sequences that is not reused in the same pool. This design means that any index-hopped read will contain a barcode pair not found in the sample sheet, allowing software demultiplexers such as Illumina’s BCL-Convert or Dragen® to discard the read automatically.
By contrast, combinatorial dual indexing reuses individual i7 and i5 indexes across multiple samples. In these cases, a hopped read may carry a valid, yet incorrect, barcode pair and be incorrectly assigned during demultiplexing. The use of UDIs eliminates this vulnerability by ensuring each index pair is unique and mapped to a single, known sample.
Recent benchmarking data have shown that the use of UDI can reduce sample cross-contamination from 1% or more down to below 0.01% even on ExAmp-based systems. This represents a 100-fold improvement in data integrity for sensitive multiplexed applications.
When UDIs are most critical
UDIs are especially critical in workflows that depend on accurate quantification or sample purity. This includes targeted sequencing panels with low variant allele frequencies, liquid biopsy applications, and single-cell RNA-seq. These workflows have low error tolerances and are highly vulnerable to even minor cross-contamination.
Even for discovery-focused bulk RNA-seq or exome sequencing projects, the adoption of UDIs is increasingly recommended. With commercially available kits offering 384, 768, or even 1,536 unique barcode pairs , scalability is no longer a barrier. Additionally, UDIs are compatible with most Illumina software tools and require no change to library preparation workflows aside from index selection.
While smaller-scale projects on low-throughput platforms like the MiSeq® may tolerate combinatorial dual indexing, the downward pressure on sequencing costs and rising complexity of pooled runs make UDI a safer and more future-proof choice.
Conclusion
Index hopping remains one of the more with persistent sources of cross-sample contamination in Illumina sequencing workflows. Although its frequency can be modest, its impact is magnified in multiplexed or precision experiments. By adopting unique dual indexing, researchers can confidently eliminate the vast majority of index misassignments, preserving data quality and minimizing the risk of false discoveries.
For any lab aiming to scale throughput, transition into regulated applications, or simply reduce sequencing error, UDIs represent a simple and highly effective upgrade. As NGS moves into more sensitive and higher-plex domains, best practices like unique dual indexing will become foundational to trustworthy genomics.
References
- Illumina. (n.d.). Index Hopping. Retrieved from https://www.illumina.com/techniques/sequencing/ngs-library-prep/multiplexing/index-hopping.html
- van der Valk, T. et al. Genome Biology 24, 215 (2023). DOI:10.1186/s13059-023-02979-9
- Illumina. (n.d.). Understanding unique dual indexes (UDI) and associated library prep kits. Retrieved from https://knowledge.illumina.com/library-preparation/general/library-preparation-general-reference_material-list/000002344



